When you take a pill once a day instead of three times, it’s not magic-it’s science. Modified-release (MR) formulations are engineered to release medication slowly over time, smoothing out blood levels and cutting down on side effects. But getting these generics approved isn’t as simple as copying an immediate-release tablet. There’s a whole different set of rules, and if they’re not followed exactly, the drug might work differently-even if it looks identical.
Why Modified-Release Drugs Need Special Rules
Immediate-release pills dump their drug into your bloodstream fast. Modified-release ones? They’re like slow-burning engines. Extended-release (ER) tablets, delayed-release capsules, or multiphasic systems (like Ambien CR) release drug in stages. That means their bioequivalence can’t be judged just by peak concentration and total exposure. You have to see when and how the drug comes out.
The FDA, EMA, and WHO all agree MR drugs need stricter testing than regular pills. Why? Because a small change in the coating, bead size, or polymer matrix can shift the release pattern. That shift might not show up in total drug exposure (AUC), but it could cause dangerous dips or spikes in blood levels. For drugs like warfarin or antiepileptics, even a 10% difference in early release can mean the difference between control and a seizure.
Key Metrics That Matter
For immediate-release drugs, you only need to check two things: AUC (total exposure) and Cmax (peak level). For MR drugs, that’s not enough.
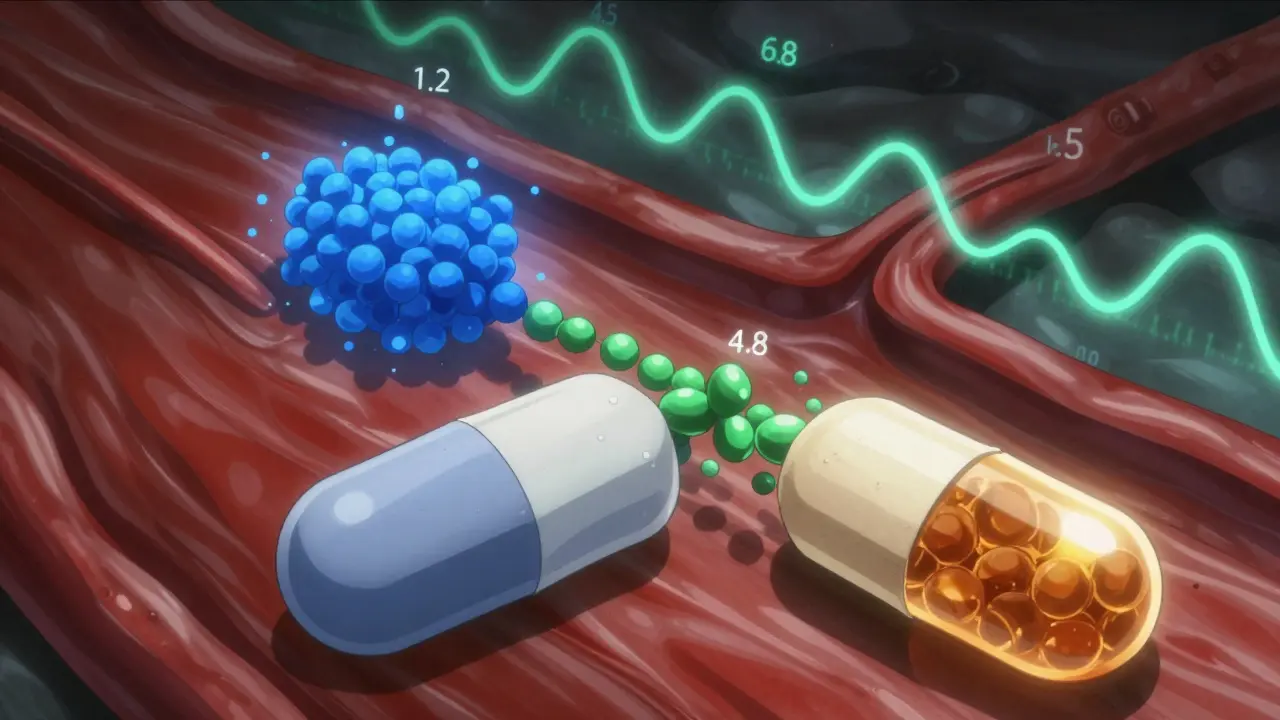
- Partial AUC (pAUC): This measures drug exposure during specific time windows. For example, Ambien CR has two phases: an immediate-release hit at 0-1.5 hours, then a slow release after. Both parts must match the brand within 80-125%. If the early phase is too weak, you won’t fall asleep fast enough. Too strong? You might feel groggy in the morning.
- Dissolution profiles: Regulators don’t just test how much drug comes out-they test how it comes out. ER tablets must dissolve in three pH levels: stomach acid (pH 1.2), small intestine (pH 4.5), and colon (pH 6.8). The similarity score (f2) must be above 50. If your generic dissolves too fast in acid, it could cause stomach upset or dose dumping.
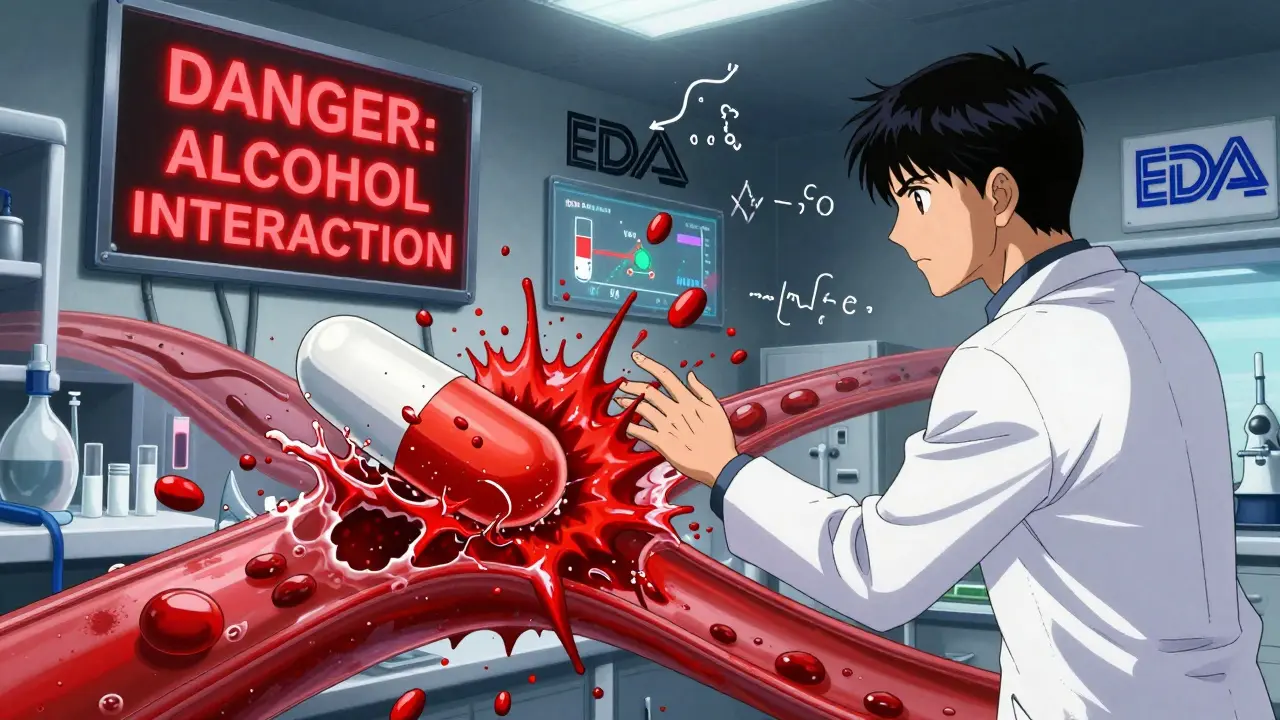
- Alcohol interaction tests: If your ER pill contains 250 mg or more of active ingredient, it must be tested in 40% ethanol. Alcohol can break down some coatings, causing the whole dose to flood into your system at once. Between 2005 and 2015, seven ER products were pulled off the market for this exact reason.
Regulatory Differences Between Agencies
The FDA and EMA don’t always see eye to eye on MR bioequivalence-and that trips up manufacturers.
The FDA says: One dose, fasted, is enough. They believe single-dose studies are more sensitive to formulation differences. Since 2015, 92% of approved ER generics used this method. The EMA, however, still requires steady-state studies for drugs that build up in the body (accumulation ratio >1.5). That means patients take the drug daily for days or weeks before testing. It’s more expensive and harder to run.
For multiphasic drugs, the FDA demands pAUC at exact timepoints. The EMA prefers half-value duration (HVD) and midpoint duration time (MDT)-metrics that describe how long the drug stays active, not when it peaks. This difference has caused delays in approvals. A generic for Concerta (methylphenidate ER) was rejected in 2012 because it didn’t match the brand’s early release profile, even though total AUC was fine.
Special Cases: High Variability and Narrow Therapeutic Index
Some drugs are naturally unpredictable in the body. Warfarin, tacrolimus, and dextroamphetamine are examples. Their within-subject variability exceeds 30%. For these, standard 80-125% bioequivalence limits don’t work.
That’s where Reference-Scaled Average Bioequivalence (RSABE) comes in. Instead of a fixed range, the acceptable window expands based on how variable the reference product is. But there’s a cap: the upper limit can’t exceed 57.38% of the reference’s variability. This method is now mandatory for highly variable MR drugs under FDA guidance (2018).
For drugs with a narrow therapeutic index (NTI)-where small changes can be dangerous-the FDA tightens the rules even further. Acceptance limits shrink to 90.00-111.11%. And you must prove your generic has the same variability as the brand. No shortcuts.
Real-World Failures and Wins
Developing an MR generic is expensive and risky. A typical study costs $1.2-1.8 million-$500,000 more than an immediate-release version.
One Teva scientist reported that 35-40% of early ER oxycodone formulations failed dissolution testing at pH 1.2. The coating would dissolve too fast in stomach acid. It took six months of reformulation to fix.
On the flip side, Sandoz got approval for an ER tacrolimus generic using a biowaiver-no human study needed. How? Their dissolution profile matched the brand perfectly at pH 6.8 (f2=68). That saved $1.5 million and 10 months.
But failures still happen. In 2021, 22% of MR generic applications were rejected by the FDA because they didn’t properly assess pAUC. Many companies assumed AUC and Cmax were enough. They weren’t.

What It Takes to Get It Right
There’s no way around it: MR bioequivalence needs expertise.
- You need dissolution testers that can handle complex profiles-USP Apparatus 3 or 4, not the standard paddle (Apparatus 2).
- You need pharmacokinetic modeling tools like Phoenix WinNonlin or NONMEM to calculate pAUC and RSABE.
- You need to know the product-specific guidance (PSG) for your drug. The FDA has over 150 PSGs for MR products. Missing one can kill your application.
Training takes time. According to a 2022 survey, it takes 12-18 months for a scientist to become proficient in MR BE testing. Most small biotechs can’t afford this. That’s why 97% of MR generic applications come from big pharma or large CROs like Covance or ICON.
The Future Is Here
Regulators are catching up. The EMA is drafting new guidelines that will likely drop the steady-state requirement for most MR drugs, aligning more with the FDA. That’s good news for manufacturers.
More companies are using in vitro-in vivo correlation (IVIVC) models. If you can prove mathematically that your tablet’s dissolution in a lab predicts how it behaves in the body, you might skip human studies entirely. The FDA has accepted this for 12 products since 2019, including paliperidone ER.
And soon, mechanistic absorption models (PBPK) will play a bigger role. These simulate how the drug moves through the gut, liver, and bloodstream. By 2024, the FDA plans to release new guidance on complex MR systems like gastroretentive tablets and multiparticulate beads.
The bottom line? Modified-release drugs are growing fast. By 2028, they’ll make up 42% of all prescription sales. But with great benefit comes great complexity. If you’re developing a generic MR product, you’re not just copying a pill-you’re recreating a delivery system. And that takes precision, patience, and a deep understanding of the science behind it.
Why can’t we use the same bioequivalence rules for modified-release and immediate-release drugs?
Immediate-release drugs release all their content quickly, so measuring total exposure (AUC) and peak level (Cmax) is enough. Modified-release drugs are designed to release slowly or in stages. If you only check AUC and Cmax, you might miss critical differences in how the drug enters the bloodstream-like a delayed start, a second peak, or too-fast release. That’s why partial AUC, dissolution profiles, and alcohol testing are required for MR products.
What is pAUC and why is it important?
pAUC stands for partial area under the curve. It measures drug exposure during a specific time window-like the first 1.5 hours after taking a pill. For multiphasic drugs like Ambien CR, the early phase helps you fall asleep, and the later phase keeps you asleep. If the generic releases too little drug in that early window, it won’t work as well. Regulators require pAUC to be within 80-125% of the brand to ensure therapeutic equivalence.
Do alcohol tests really matter for extended-release pills?
Yes. Alcohol can break down the polymer coatings or matrixes in ER tablets, causing the entire dose to be released at once. This is called dose dumping. Between 2005 and 2015, seven ER drugs were withdrawn because patients who drank alcohol had dangerous spikes in blood levels. Now, any ER product with 250 mg or more of active ingredient must be tested in 40% ethanol. It’s not optional-it’s a safety requirement.
What’s the difference between FDA and EMA guidelines for MR bioequivalence?
The FDA mostly uses single-dose, fasting studies and focuses on pAUC and dissolution profiles. The EMA sometimes requires multiple-dose (steady-state) studies, especially if the drug builds up in the body. The EMA also uses metrics like half-value duration (HVD) instead of pAUC. These differences have caused delays in global approvals, but the EMA is moving toward aligning with the FDA’s approach.
Why are MR generics more expensive to develop than immediate-release ones?
MR generics require more complex studies: multiple dissolution conditions, alcohol testing, pAUC calculations, and sometimes RSABE modeling. A single-dose MR bioequivalence study costs $1.2-1.8 million, compared to $0.8-1.2 million for immediate-release. Plus, failure rates are higher-up to 40% in early development-meaning more rounds of reformulation. That adds up fast.
Can a generic MR drug be approved without human studies?
Yes, but only under strict conditions. If a manufacturer can prove perfect similarity in dissolution profiles across multiple pH levels (f2 ≥ 50), and the drug meets biowaiver criteria (like low solubility and high permeability), regulators may waive human studies. Sandoz did this successfully with an ER tacrolimus product. But this only works for a limited set of drugs and formulations.
What happens if a generic MR drug passes bioequivalence but still causes side effects?
That’s a known risk. Some studies, including one in Neurology (2016), found that 18% of generic extended-release antiepileptic drugs had higher seizure breakthrough rates than the brand, even though they met all bioequivalence criteria. This suggests that current tests might not capture subtle differences in release patterns that affect real-world outcomes. Regulators are aware and are working on better models, like PBPK, to predict these effects.
Author

Lana Kabulova
January 21, 2026 AT 15:02arun mehta
January 23, 2026 AT 11:48Chiraghuddin Qureshi
January 24, 2026 AT 14:33Tatiana Bandurina
January 25, 2026 AT 08:02Philip House
January 26, 2026 AT 22:58Akriti Jain
January 28, 2026 AT 13:07Mike P
January 29, 2026 AT 16:36Jasmine Bryant
January 30, 2026 AT 00:20Liberty C
January 30, 2026 AT 08:58